
Thérapie Génique
Corinne Kostic et Yvan Arsenijevic
Thérapie génique par AAV de la rétinite pigmentaire 28 (RP28) associée au gène FAM161A
La rétinite pigmentaire (RP) est une maladie dégénérative de la rétine dont l’incidence est de 1:4000 dans le monde. Cette dystrophie des bâtonnets et de cônes évolue lentement vers la cécité et aucun traitement efficace n’est actuellement disponible. Parmi les cas de RP, la RP28 est une ciliopathie affectant uniquement l’œil qui a été associée à des mutations récessives du gène humain FAM161A dans plusieurs populations ethniques (Bandah-Rozenfeld et al. 2010, Langmann et al. 2010). L’expression de la protéine FAM161A a été observée dans le cil connecteur du photorécepteur (réf) et nous avons ensuite démontré, en collaboration avec le laboratoire de Paul Guichard et Virginie Hamel (UNIGE), que la protéine FAM161A appartient à la structure d’échafaudage interne du cil connecteur du photorécepteur de souris (Mercey et al. 2022). La souris déficiente pour Fam161Atmb/tmbmise au point par nos collègues Avigail Beryozkin et Dror Sharon (Hadassah Medical Center) reproduit la progression de la maladie humaine (Beryozkin et al. 2021). Nous avons démontré l’amélioration de la survie et de la fonction des photorécepteurs dans le modèle de souris Fam161Atmb/tmbpar l’administration sous-rétinienne d’un vecteur de virus adéno-associé (AAV), qui code pour l’ADNc long de Fam161A (Matsevich et al. 2023). En transposant cette approche avec le gène FAM161A humain, il a été observé que seule la co-injection de deux vecteurs AAVs, qui délivrent à la fois les isoformes longue et courte humaines, permettait de sauver la fonction rétinienne de la souris Fam161Atmb/tmb. En outre, un niveau d’expression approprié est nécessaire, ce que nous avons obtenu grâce à l’utilisation d’une nouvelle séquence régulatrice dérivée du gène humain FAM161A (Arsenijevic et al. 2024). Nous continuons d’étudier de la fonction de FAM161A et d’évaluer des vecteurs AAV pour transférer cette stratégie de remplacement de gène vers une application clinique future.
Transfert de gène effectué par vecteur lentiviral pour les Amauroses de Leber causées par une déficience de RPE65
L’amaurose congénitale de Leber (ACL), la dystrophie rétinienne héréditaire enfantine la plus précoce et la plus sévère, est la cause principale de cécité chez les enfants. 10-15% des patients souffrant d’ACL portent une mutation du gène RPE65.
Nos chercheurs poursuivent une étude pré-clinique de transfert du gène sain RPE65 dans des modèles d’amaurose congénitale de Leber (ACL) de souris grâce à des injections sous-rétiniennes de vecteurs lentiviraux.
L’originalité de leur travail a été de définir une fenêtre thérapeutique précise ainsi que l’effet du transfert de gène RPE65 sur les cônes, la population de photorécepteurs la plus rapidement touchée dans ce modèle. Grâce à des mesures par électrorétinogramme (ERG) de bonnes réponses rétiniennes ont été obtenues dans des souris RPE65-/- traitées précocement (à 5 jours de vie) avec un vecteur lentiviral transférant le gène RPE65 (Bemelmans et al., 2006).
L’amélioration de la fonction des cônes a peu être démontrée. La sauvegarde de ces cellules a aussi été observée par des analyses histologiques.
Cependant ces mêmes cônes ne sont pas protégés quand le traitement est appliqué aux jeunes souris adultes (à 1 mois) démontrant une limite temporelle pour une préservation efficace. La moitié des patients portant une mutation RPE65 n’ont pas forcément une perte totale de la fonction de cette protéine ce qui peut avoir une conséquence sur le développement de la pathologie et donc sur la fenêtre thérapeutique.
Cette approche a été répétée sur un modèle de souris portant une mutation faux-sens retrouvée chez certains patients (R91W) (samardzija et al 2008). Dans ce modèle, la dégénérescence des cônes est retardée car il y a un reste d’expression de la protéine RPE65. Les mêmes types d’analyses cités ci-dessus ont révélé une sauvegarde partielle des cônes après un traitement à 1 mois d’âge (Kostic et al 2011). Non seulement le nombre de cônes répertoriés à l’âge du traitement est maintenu, mais 20% de plus de cônes (par rapport au nombre total dans la rétine de la souris saine) sont « rajeunis ». En effet, ce résultat suggère qu’une partie de la population des cônes n’exprime plus ses protéines spécifiques à ce stade (S-opsin, GNAT2) et ces cellules survivent comme des « fantômes » prêtes à récupérer leurs caractéristiques si un traitement adéquat est appliqué.
Nous avons ensuite évalué la sécurité d’une production de vecteurs de type GMP dans des conditions similaires à celles requises pour les essais cliniques mais avec une seule application topique de pommade dexaméthasone/oxytétracycline et sans anti-inflammatoire avant l’injection (Matet et al. 2017). Cette étude montre l’absence de détection des vecteurs en dehors du compartiment oculaire et une tolérance limitée au LV après la voie d’administration sous-rétinienne, ce qui suggère le besoin transitoire d’anti-inflammatoires pour contrecarrer les effets secondaires de l’inflammation comme cela est habituellement appliqué pour d’autres vecteurs viraux. Nous avons enfin démontré que notre vecteur peut augmenter le niveau de l’ARNm de RPE65 dans les cellules de l’épithelium pigmentaire dérivées d’iPSCs humaines (Udry et al. 2020), ce qui soutient fortement le potentiel de ce vecteur pour les applications cliniques de l’expression de gènes dans l’épithélium pigmentaire.
Développement de vecteurs lentiviraux pour la thérapie génique oculaire
La plupart des dégénérescences rétiniennes sont une conséquence du mauvais fonctionnement des photorécepteurs ou de l’épithélium pigmentaire ce qui nous a amené à définir les outils appropriés pour cibler spécifiquement ces types cellulaires.
Une base de l’approche par thérapie génique est d’atteindre le tissu ou la cellule approprié(e) de manière spécifique afin d’éviter des effets secondaires indésirables. Cette spécificité peut être déterminée par un tropisme limité du vecteur, par une fonction spécifique restreinte à un type cellulaire particulier ou par une expression spécifique déterminée par le choix du promoteur.
Cette dernière possibilité a été sélectionnée et nos chercheurs ont examiné l’activité de différents promoteurs dans la rétine murine après transfert de gène grâce à des vecteurs lentiviraux (Kostic et al., 2003). Ils ont aussi montré que la diffusion limitée de ces vecteurs dans l’espace sous-rétinien est due à une barrière physique entre les photorécepteurs et l’épithélium pigmentaire qui peut être altérée par l’utilisation de certains enzymes (Grüter et al., 2005). De plus, ils ont observé que le ciblage des vecteurs lentiviraux dépend du stade de la progression de la maladie (Calame et al 2011). Notamment les vecteurs lentiviraux pseudotypés avec l’enveloppe Mokola ciblent efficacement les cellules de Müller en cas de gliose dans le modèle murin Rho-/-. (Calame et al 2011).
Nos chercheurs poursuivent des études pour améliorer le transfert de gène dans la rétine grâce aux vecteurs lentiviraux.
Modèles pathologiques :
Un modèle de porc pour la dystrophie des cônes :
La compréhension des mécanismes de pathologie est cruciale pour les chercheurs qui veulent développer des nouvelles voies thérapeutiques. Des avancées majeures sont possibles grâce à des modèles animaux qui reproduisent des pathologies humaines. A cause des différences intrinsèques entre les organismes les limitations de ce genre de modèles doivent être prises en compte pour chaque maladie.
Dans ce contexte, les rongeurs sont une espèce limitée pour modéliser les maladies affectant les cônes chez les humains. En effet la distribution et l’organisation de ce type de photorécepteur sont adaptées à la vision de jour chez les primates tandis qu’il est adapté à la vision nocturne pour les rongeurs. Il est donc préférable d’étudier les déficiences visuelles liées aux cônes chez des animaux avec une région de la rétine enrichie en cônes et une taille d’œil similaire à l’homme. Les porcs qui ont en plus un taux élevé de reproduction conviennent donc particulièrement bien à ce genre de recherche.
La technique de transgénèse par vecteur lentiviral a été utilisée pour exprimer un allèle dominant du gène GUCY2D dans la rétine de porc et servir de modèle de la maladie CORD6. La caractérisation des porcs GUCY2DE837D/R838S a révélé un impact fonctionnel sur la vision ainsi que des modifications structurelles de la rétine. Des anormalités ont été détectées à l’âge de 11 semaines avec une progression lente dans l’année suivante. Une grande variété de sévérités a été constatée parmi les animaux porteurs de la mutation, d’une absence totale de symptôme à un grand déficit de fonction rétinienne.
Cette observation est particulièrement intéressante car elle imite la large hétérogénéité de sévérités décrites chez les patients CORD6 humains. Cette hétérogénéité trouvée chez les porcs est en accord avec la nature unique de chaque animal généré avec cette technique de vecteur lentiviral: le nombre de copies et les sites d’intégration du vecteur propres à chaque individu offrent la possibilité de créer un large échantillonnage d’animaux qui ont en commun la signature pathologique due à la mutation.
Cette diversité confirme l’effet pathologique de l’allèle et peut aider à identifier les facteurs intrinsèques et environnementaux impliqués dans la pénétrance. L’efficacité de cette technique a ainsi démontré le potentiel de cette stratégie pour générer des modèles de pathologie humaine et l’implication de la mutation de GUCY2D dans la dystrophie de cônes CORD6.
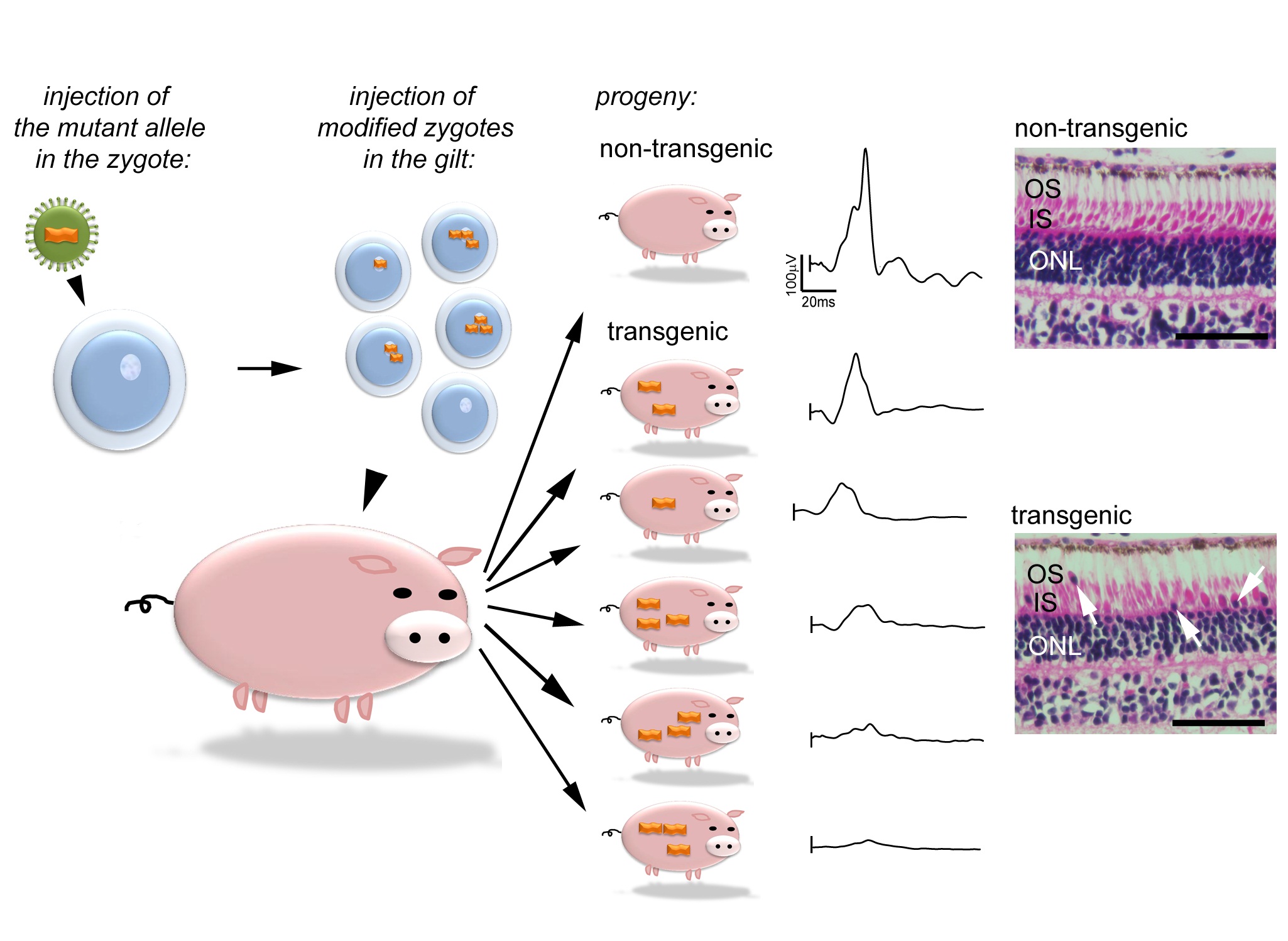
Figure – Illustration de la genèse d’un modèle de porc pour la dystrophie des cônes. Les particules de vecteur lentiviral sont injectées dans l’espace périvitellin des zygotes pour transférer l’allèle mutant (trait orange) dans le génome du porc. Les zygotes manipulés sont ensuite transférés dans l’oviducte de truies préparées qui mettent bas des individus uniques de par leur nombre de copies du transgène et du site d’intégration. La caractérisation fonctionnelle (enregistrement des réponses rétiniennes) et morphologique (histologie) des animaux confirme que le groupe présente une large variété d’altérations de la réponse rétinienne mais tous ont un nombre plus élevé de noyaux cellulaires déplacés dans la couche des segments externes (flèches) comparé aux animaux contrôles. OS: segments externes des photorécepteurs; IS: segments internes des photorécepteurs; ONL: noyaux des photorécepteurs.
